



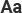
Source : Le Monde
Ce site très visité permet de suivre la propagation géographique de plusieurs épidémies virales, dont le Covid-19, presque en temps réel, à l’aide des données de séquences partagées.
L’épidémie de Covid-19 a divisé en deux la population de ceux qui tentent de la suivre. Il y a ceux qui scrutent chaque jour les chiffres du nombre de cas, de décès, d’admis en réanimation… Et il y a ceux qui regardent le site Nextstrain. Si les premiers s’intéressent aux victimes, les seconds sont concentrés sur le coupable, le coronavirus SARS-CoV-2. « Nous avons des centaines de milliers de vues quotidiennes en ce moment », indique James Hadfield, l’une des chevilles ouvrières de cette plate-forme née en 2016. Il travaille dans l’un des deux groupes qui l’a lancée, l’équipe de Trevor Bedford, au centre de recherche contre le cancer Fred-Hutchinson (Seattle, Etats-Unis). L’autre groupe est à l’université de Bâle, en Suisse, dans l’équipe de Richard Neher.
Le site parvient à synthétiser de façon très visuelle et esthétique les propriétés de plus de 3 900 génomes du nouveau virus, prélevés dans une soixantaine de pays (au 16 avril). Une carte mondiale montre l’origine géographique de ces séquences – surtout en Chine, aux Etats-Unis et en Europe – ainsi que les voies que le virus a suivies. Apparaissent également les régions de la séquence génétique où des mutations (ou substitutions) ont été notées, parmi les 30 000 « lettres » que compte ce génome.
Enfin s’affiche l’arbre généalogique du coronavirus, avec son tronc, ses branches, ses rameaux et toutes les feuilles que constituent les variants. En filtrant par pays, les quelque 200 échantillons français permettent de voir d’un seul coup d’œil qu’il y a eu probablement plusieurs introductions dans le pays, en provenance de Chine, d’Europe voire des Etats-Unis.
Une plus grande ouverture de la recherche
L’un des points forts de Nextstrain est justement de présenter ces fameux arbres qui, à la différence de l’origine géographique, sont difficiles à obtenir. Ils sont calculés par des algorithmes proposant les relations les plus probables entre ces séquences qui, parfois, ne varient que de quelques lettres. A partir de là, les chercheurs traquent l’origine de la contamination, surveillent des mutations dangereuses, voire en déduisent des propriétés de la transmission de l’épidémie. « Nous voulions présenter ces analyses en temps réel de façon à ce qu’elles servent lors d’une épidémie. La manière traditionnelle de publier en science n’est pas adaptée dans ce cas-là, car les résultats peuvent ne pas être à jour. Evidemment, Nextstrain ne veut pas remplacer ces articles », explique James Hadfield.
Pour Nextstrain, tout a commencé en 2016, avec des analyses sur l’épidémie due au virus Ebola en Afrique de l’Ouest et avec d’autres travaux, rétrospectifs ceux-là, sur le virus du Nil occidental. Ont suivi la grippe A (H1N1), la tuberculose, la dengue… Avec le coronavirus, le site a changé de dimension avec plus de 3 900 séquences puisées dans la base de données Gisaid, gérée par le gouvernement allemand et une association. « Le plus dur du travail est de se tenir à jour ! », souligne James Hadfield.
L’initiative témoigne aussi d’une plus grande ouverture de la recherche. Les séquences sont partagées, les logiciels utilisés ont des codes sources ouverts, les traductions de Nextstrain (en 17 langues) sont assurées par des volontaires, pas forcément biologistes. Un souci pédagogique confirmé par les tutoriels et les résumés de ce que toutes ces séquences nous apprennent de l’épidémie.
Comme les programmes sont librement accessibles, les chercheurs les utilisent aussi dans leurs laboratoires pour leurs propres calculs et visualisations. « Ma satisfaction est qu’en ce moment nos outils aident l’Institut national de recherche biomédicale de la République démocratique du Congo à faire face à l’épidémie d’Ebola », se réjouit James Hadfield.
Source : Le Monde
Source : Sciences et Avenir
A partir des données génomiques recueillies sur des milliers d’échantillons du virus SARS-CoV-2, la plateforme Nextstrain permet de reconstruire et de visualiser la chronologie et la diffusion spatiale de l’épidémie de Covid-19. Décryptage avec Samuel Alizon, directeur de recherche CNRS, spécialiste de la biologie évolutive des agents pathogènes.
Un virus ça mute un peu tout le temps. C’est même grâce à ça que la phylodynamie retrouve sa trace. Chaque fois qu’une particule virale (virion) se réplique dans une cellule, elle doit recopier son code génétique, une succession de 30.000 bases nucléiques de l’ARN, représentées par les lettres A, G, U, C. Mais ce système commet souvent des erreurs, remplaçant une lettre par une autre : ce sont les mutations. « Ce sont autant de traces qui s’accumulent dans les génomes du virus qui permettent de reconstituer la structure de l’épidémie, sa chronologie et sa diffusion spatiale », explique Samuel Alizon, directeur de recherche CNRS/IRD, spécialiste de l’écologie et de l’évolution des agents pahogènes. Il faut imaginer les milliards de milliards de virions du SARS-CoV-2 qui circulent parmi les humains comme une gigantesque famille, dans laquelle les mutations se transmettent de proche en proche. « Il s’agit d’établir des liens de parentés entre les échantillons séquencés. Plus leurs séquences se ressemblent, plus elles sont proches. Cela permet de reconstituer une sorte d’arbre généalogique : la phylogénie du virus », détaille Samuel Alizon (voir image ci-dessous).
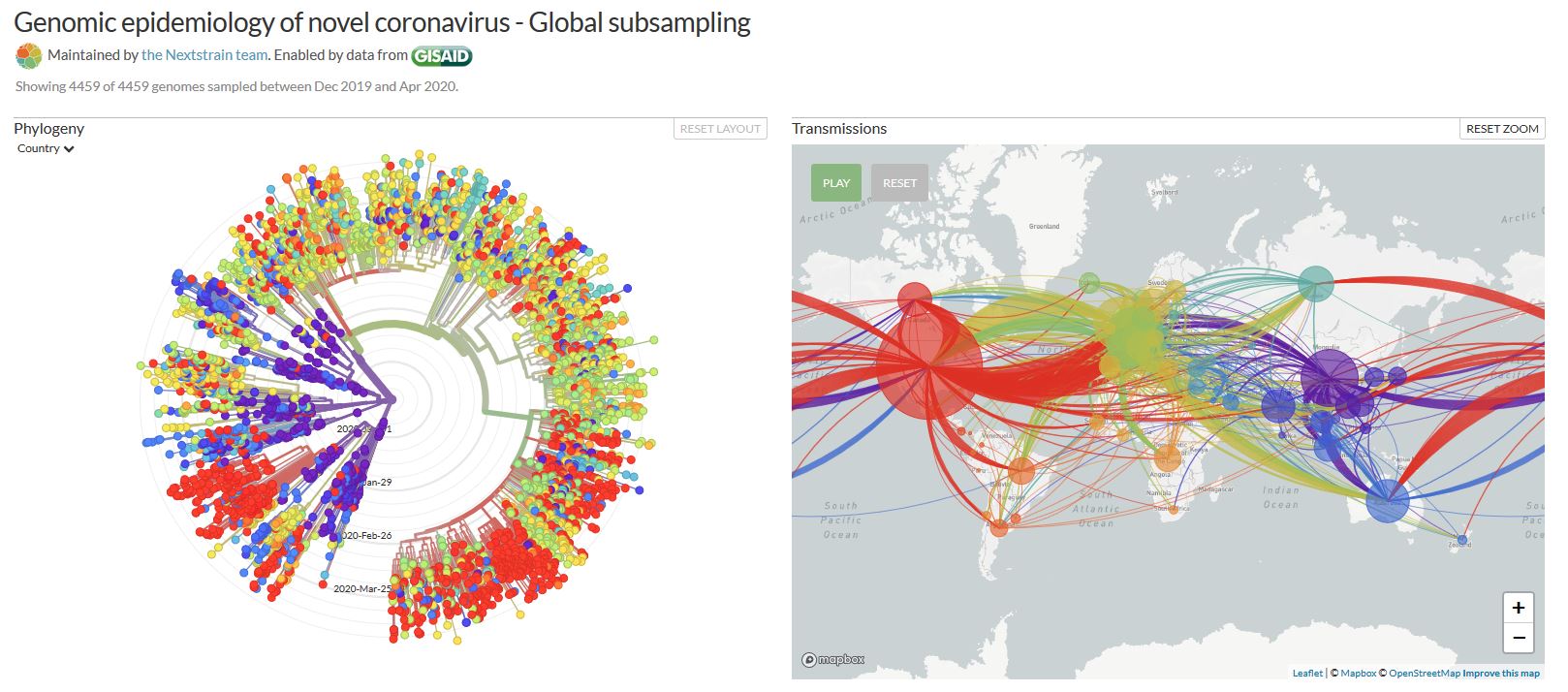
La capture d’écran de la plateforme Nextstrain ci-dessus montre l’arbre phylogénétique du virus SARS-CoV-2. Chaque point représente ainsi l’une des 3425 séquences génétiques réalisées à partir d’échantillons prélevés chez des personnes infectées. Chaque couleur représente le pays où le virus a été prélevé et séquencé : la Chine en violet, les Etats-Unis en rouge, le Royaume-Uni en jaune, les pays de l’Union européenne en vert, etc. En tout, 57 pays sont inscrits, avec une surreprésentation des pays qui ont les plus fortes capacités de test et de séquençage.
« Nextstrain est un outil collaboratif de visualisation de l’information des génomes viraux en temps presque réel puisqu’il est implémenté au fur et à mesure. Ça sert d’abord à nettoyer les données génétiques, puis à établir la phylogénie du virus grâce à des modèles statistiques qui traitent les jeux de séquences. Ce sont deux étapes très robustes », précise Samuel Alizon. « Ça permet ensuite de dater les événements de façon assez puissante. La date du séquençage est une aide, certes, mais elle ne suffit pas à estimer quand ont été infectées les personnes, ni à qui elle a eu le temps de le transmettre, poursuit le chercheur. Enfin, l’analyse va reconstituer la diffusion spatiale de l’épidémie, avec une moindre fiabilité toutefois. »
Comme outil de visualisation, la plateforme Nextstrain est relativement accessible au néophyte pour explorer cette phylogénie, avec les dates approximatives d’introduction du virus dans tel ou tel pays par exemple. Plus parlant encore, une animation présente l’évolution géographique et phylogénique simultanée de l’épidémie, replacée dans sa chronologie. La vidéo ci-dessous en donne un aperçu (l’intervalle des dates apparaît dans la colonne de gauche, au centre l’arbre phylogénique, à droite la diffusion géographique) :
Que nous apprend la phylodynamique sur l’épidémie de Covid-19 ?
Avec son équipe de l’institut MiVEGEC (Maladie infectieuses et vecteurs écologie, génétique, évolution et contrôle), Samuel Alizon a publié un rapport sur la phylodynamique du Covid-19 en France à partir des 69 séquences disponibles. « Le principal enseignement, c’est qu’il y a vraisemblablement eu plusieurs introductions en France à différents moments. D’abord, les cas venus directement de Chine repérés entre le 23 et le 29 janvier (3 séquences). Assez vite identifiés et isolés, ces derniers n’ont semble-t-il pas eu de descendance. « On constate aussi que la majorité des séquences françaises sont regroupées dans le clade supérieur de la phylogénie (voir ci-dessous) et que les séquences récoltées le plus tôt dans l’épidémie (donc sur la gauche du graphique) sont dans des clades moins massifs, qui ont donc causé moins d’infections secondaires », écrivent les chercheurs.
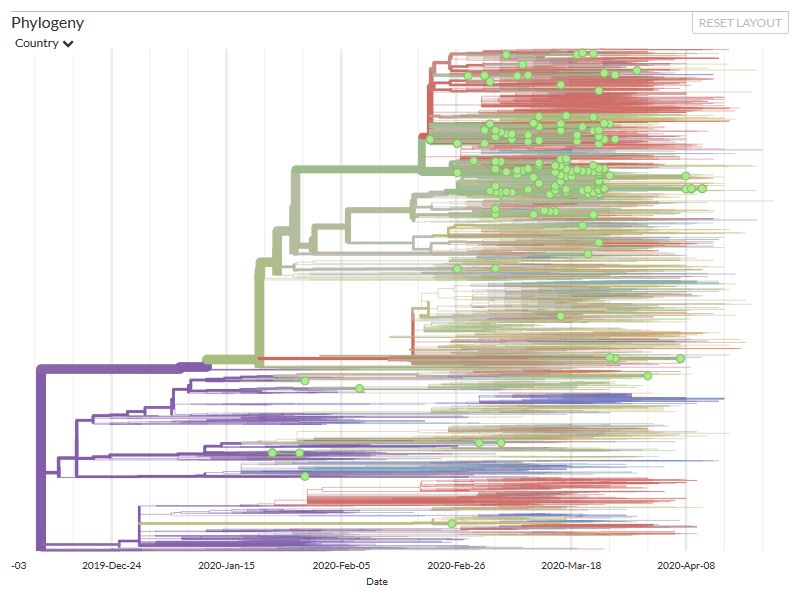
L’origine des deux clades principaux (en vert) est attribuée à un ancêtre commun au Royaume-Uni par Nextstrain. Samuel Alizon prévient toutefois qu’il s’agit là d’un « résultat sensible à la quantité d’échantillonnage. Or le Royaume-Uni a quatre fois plus séquencé que la France. L’ancêtre commun pourrait donc provenir d’un autre pays d’Europe voire de Chine. » En revanche, l’examen des dates donne des résultats plus solides, et assez inquiétants : « Le clade principal en France qui regroupe 59 des 68 séquences disponibles a commencé à croître après le 18 janvier et avant le 13 février (nextstrain le date lui au 25 janvier). Or, le premier cas importé détecté date du 24 janvier et la courbe du nombre de nouveaux cas par jour n’a commencé croître que le 27 février. Autrement dit, avec une erreur minime, on peut dire que l’épidémie était déjà installée en France ou proche de la France deux semaines avant la détection des premiers cas. »
« On ne peut pas exclure qu’il y ait eu encore d’autres introductions du virus qui n’apparaissent pas avec les échantillons séquencés », rappelle Samuel Alizon. Face à une pandémie aux proportions du Covid-19, la phylodynamique se heurte en effet a une faible densité d’échantillonage, soit le nombre de séquences disponibles selon les pays. Mais l’examen de la phylogénie montre par exemple qu’une séquence française du 25 février pourrait descendre d’une branche espagnole.
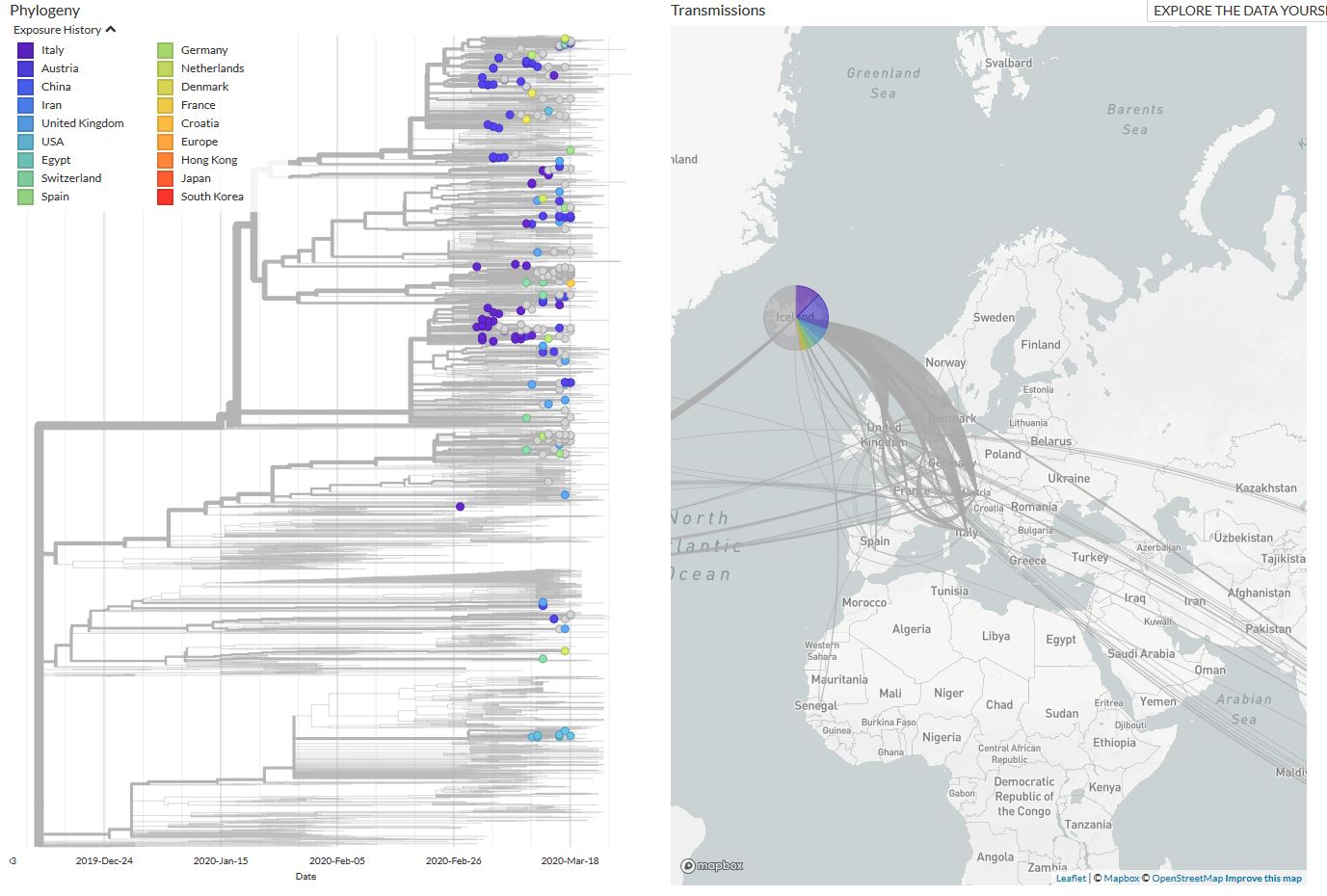
Enfin, le rapport officiel de Nextstrain du 27 mars par l’équipe de Trevor Bedford observe « un fort mélange d’échantillons à travers l’Europe, ce qui laisse penser que le virus a continué à traverser les frontières au cours des 3 à 5 dernières semaines. Lorsque les mesures d’atténuation auront eu le temps de faire sentir leurs effets, il se peut que nous constations un plus grand nombre de cas groupés par pays. » Ce n’était toujours pas le cas le 3 avril, date du dernier rapport. Les données venues d’Islande, pays qui a beaucoup testé et séquencé, montre qu’environ la moitié des infections ont été contractées en dehors de l’Islande, tandis que la moitié sont présumées être des infections contractées localement (en gris sur la carte ci-dessous).
En Amérique du Nord, l’introduction du virus est encore plus précoce qu’en Europe. La première séquence est issue d’une lignée chinoise, et provient d’un échantillon prélevé le 19 janvier dans le comté de Snohomish, au nord de Seattle, dans l’Etat de Washington. C’est là que l’épidémie s’est répandue en premier. « Mais alors que beaucoup des premiers cas étaient associés à des voyages, les cas plus récents, provenant de nombreux États différents, sont largement mêlés les uns aux autres de part et d’autre de l’arbre, explique le dernier rapport Nextstrain. Il est clair que ces foyers localisés sont le résultat d’un mélange intensif. »
Les origines du virus
« L’un des résultats les plus importants de la phylogénie, c’est d’avoir réussi à dater l’origine de l’épidémie et de garantir qu’il n’y a eu qu’une seule introduction, estimée entre le mois d’août et la fin novembre 2019 », rappelle Samuel Alizon. « En analysant la structure des mutations sur des coronavirus proches et en modélisant leur effet en terme d’infectiosité, nous connaissions tous les ingrédients permettant de rendre des souches portées par des chauves-souris contagieuses pour l’humain », explique le chercheur. « La surprise, c’est que ce sont des mutations que nous n’avions pas envisagées qui ont permis à SARS-CoV-2 de sauter la barrière de l’espèce. » D’un côté une souche connue, hébergée par des chauves-souris est quasiment identique à SARS-CoV-2. Sauf que sa protéine de surface Spike, dont le virus se sert pour infecter les cellules, n’est pas équipée pour s’attacher aux cellules humaines. De l’autre côté, la souche de pangolin dont on a beaucoup entendu parler ressemble un peu moins au virus humain, mais présente les mutations nécessaires pour équiper Spike contre les humains. « Il est toujours possible de s’imaginer une fuite du fameux laboratoire P4 de Wuhan. Ce sont des choses qui peuvent arriver. Mais en l’occurrence c’est assez improbable et, de toute façon, on ne saura jamais. Si le virus avait été fabriqué en revanche, il aurait été équipé des mutations déjà connues pour ne pas attirer l’attention », analyse Samuel Alizon.
Finalement, la phylodynamique se fixe des objectifs très proches de ceux de l’épidémiologie classique qui se fonde sur des données d’incidence, c’est-à-dire les cas d’infection. « Cette discipline relativement récente propose d’estimer des valeurs de paramètres d’intérêt épidémiologique à partir de données de séquences génétiques et à l’aide de modèle statistiques. On peut ainsi estimer le taux de reproduction de l’épidémie ». Le fameux R0 qui correspond au nombre moyen de personnes contaminées par un malade. Ce qu’on cherche à obtenir, c’est une phylogénie d’infection. » Autrement dit, faire coïncider l’analyse génomique avec les données d’incidence. « Dans un contexte d’échantillonage total, où l’on séquencerait le virus chez tous les malades d’une épidémie, il serait ainsi possible de reconstituer tout l’arbre de transmission d’une épidémie. » Qui infecte qui, où, quand et comment ? Mais cette « épidémiologie génomique » est encore très peu intégrée à la santé publique en France. « L’idéal serait de pouvoir combiner les deux approches, mais ça pose des problèmes mathématiques très complexes pour rendre les données interropérables », explique Samuel Alizon dont l’équipe vient de déposer un projet pour tenter de réduire ce fossé.
Source : Sciences et Avenir
![]()
Nous vous proposons cet article afin d'élargir votre champ de réflexion. Cela ne signifie pas forcément que nous approuvions la vision développée ici. Dans tous les cas, notre responsabilité s'arrête aux propos que nous reportons ici. [Lire plus]Nous ne sommes nullement engagés par les propos que l'auteur aurait pu tenir par ailleurs - et encore moins par ceux qu'il pourrait tenir dans le futur. Merci cependant de nous signaler par le formulaire de contact toute information concernant l'auteur qui pourrait nuire à sa réputation.









Commentaire recommandé
» Or, le premier cas importé détecté date du 24 janvier et la courbe du nombre de nouveaux cas par jour n’a commencé croître que le 27 février. Autrement dit, avec une erreur minime, on peut dire que l’épidémie était déjà installée en France ou proche de la France deux semaines avant la détection des premiers cas. »
Mais alors que faire de cette communication ? :
https://francais.rt.com/france/74777-covid-19-cas-teste-positif-fin-decembre-selon-chef-service-reanimation
15 réactions et commentaires
» Or, le premier cas importé détecté date du 24 janvier et la courbe du nombre de nouveaux cas par jour n’a commencé croître que le 27 février. Autrement dit, avec une erreur minime, on peut dire que l’épidémie était déjà installée en France ou proche de la France deux semaines avant la détection des premiers cas. »
Mais alors que faire de cette communication ? :
https://francais.rt.com/france/74777-covid-19-cas-teste-positif-fin-decembre-selon-chef-service-reanimation
+13
AlerterDit dans l’article : « On ne peut pas exclure qu’il y ait eu encore d’autres introductions du virus qui n’apparaissent pas avec les échantillons séquencés », rappelle Samuel Alizon.
+4
Alerterje pense qu’il faut lire « au moins deux semaines »
Il serait intéressant de voir où ce premier cas se situe dans la phylogénie proposée dans l’article.
+0
Alerterhttp://www.leparisien.fr/amp/international/covid-19-des-athletes-francais-contamines-a-wuhan-en-octobre-05-05-2020-8311221.php
Il va falloir étirer la figure à gauche…
+3
AlerterL’idée est intéressante. Mais les faux positifs existent… https://la1ere.francetvinfo.fr/nouvellecaledonie/tests-covid-19-un-faux-positif-et-un-faux-negatif-confirmes-par-le-cht-819510.html
Effectivement, c’est intrigant. Il faudrait avoir plus d’informations, malheureusement cet article de journaliste (?) est trop sommaire pour en avoir une vraie idée…
+0
Alerterhttps://www.dna.fr/sante/2020/05/07/l-hopital-de-colmar-evoque-des-cas-de-covid-des-novembre
Novembre examen scanners
+0
AlerterOui, des athlètes français des jeux militaires d’octobre à Wuhan qui se sont plaints de symtômes du Covid, ce qui corroborrerait la thèse que le porte parole du gouvernement chinois avait fait valoir, à savoir que des soldats US avaient importé le virus… (à mettre en relation avec les cas de Covid avérés par le directeur du CDC, et la fermeture du laboratoire P4 de Fort Detrick en août 2019… :http://www.leparisien.fr/international/covid-19-des-athletes-francais-contamines-a-wuhan-en-octobre-05-05-2020-8311221.php
+1
Alertertiens, vous avez des privilèges ? quand je dis ça je me fais modérer, tout comme je parlais du patient de colmar du 3 décembre…. (pourtant, ça fait plus d’un mois que j’ai passé les liens en mode contact)
aujourd’hui dans les news covidé, ils parlent maintenant de mi-novembre….. decodex, decodex
+0
AlerterJe retiens l’argument ‘scientifique’ qui fonde la ‘certitude’ que le virus n’a pas été fabriqué par l’homme. Il est très faible : « Si le virus avait été fabriqué en revanche, il aurait été équipé des mutations déjà connues pour ne pas attirer l’attention ». Autrement dit, ce n’est pas un marqueur non manipulable, mais un marqueur dont on imagine que le manipulateur n’a pas intérêt à le manipuler… On tombe donc dans le dilemme bien connu du gardien de but au moment du penalty : « il sait que j’arrête mieux à droite, donc il va tirer à gauche. Mais il sait que je sais qu’il sait que j’arrête mieux à droite et que je vais plonger à gauche, donc il va tirer à droite. Mais il sait que je sais qu’il sait que je sais….. » Le virologue use d’un argument psychologique, domaine qui n’a pas l’air d’être son fort. Et il exclut a priori l’hypothèse d’un docteur Frankestein un peu malin, ou au contraire un peu con : ce qui est en soi déjà un peu con. En conclusion : il n’y a pas de preuve indubitable, matériellement non-manipulable, qu’un virus est naturel. Juste des présomptions.
+7
AlerterBien que j’ai été convaincu par la thèse de la zoonose, je suis content de lire ce genre de commentaire. Le simple fait de rappeller les bases élémentaires de la logique. C’est assez effrayant parfois de voir des gens se disant scientifiques faire des affirmations de ce genre : « Rien ne prouve A, alors A est faux ». C’est une abération, une erreur élémentaire !
+2
AlerterVisiblement Nexstrain pointe la Chine comme la région du monde de départ du virus. Ce qui entre en contradiction avec des études précédentes. En outre un virus chimérique à base de coronavirus a déjà été créé aux USA en 2015. Pour retrouver le patient zéro, on ferait bien de s’intéresser aux cas de pneumonies « atypiques » dès la fin de l’été en Italie et surtout aux USA (mises sur le compte du vapotage, puis de la grippe).
+8
AlerterLes conclusions de Nextstrain me semblent parfaitement logiques vu qu’elles sont basées sur des échantillons de virus qu’on a commencé à échantillonner fin décembre/début janvier.
Si jamais le virus venait d’autre part sans que personne n’ait remarqué qu’il s’agissait d’un virus nouveau (vous citez les usa) Nexstrain ne pourrait pas le savoir… Sans échantillons.
Donc il faut reformuler l’article en commençant par « En l’état actuel de nos connaissances et sachant que notre base de données est nécessairement incomplète…… »
+10
AlerterToute la science n’est qu' »en l’état actuel de nos connaissances ».
Mais contrairement à ce que vous dites, si le virus était apparu – par exemple – en Islande, alors le site le verrait tout de suite, car c’est en Islande qu’il y aurait la plus grande diversité génétique. Mais c’est en Chine que celle-ci est maximum…
+0
Alerter«Ce qui entre en contradiction avec des études précédentes»
que vous voudrez bien relater ici ?
+0
AlerterLa phylogénie, pour l’instant, n’est compatible qu’avec une origine chinoise du virus à l’exclusion de toutes les autres. La Chine est par exemple l’unique endroit apparaissant sur toutes les branches les plus basales de l’arbre, et généralement à une position proche de la racine. Du point de vue mathématique, c’est un argument très fort en faveur de l’hypothèse chinoise. Les horloges génétiques donnent un least commun ancestor vers novembre-décembre pour tous les virus actuels.
+0
AlerterLes commentaires sont fermés.